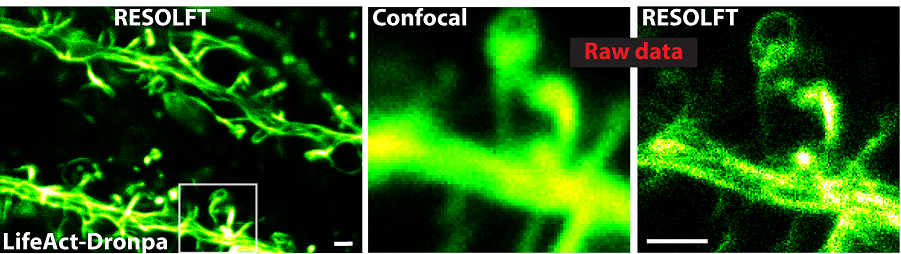
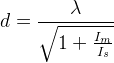RESOLFT
Reversible saturable optical fluorescence transitions
RESOLFT super resolution microscopy represents a powerful
tool to decipher spatio-temporal information coded in the life sciences by (1) minimizing the illumination light
intensities (2) employing genetically encoded markers (3) improving the spatial resolution of conventional fluorescence
microscopy down to the nanoscale. In RESOLFT nanoscopy the spatial resolution is improved by taking advantages of
“long-lived” (µs-ms) molecular states, which are populated/depopulated with light intensities in the range of W–kW/cm2 .
The precious states are so far provided by reversible switchable fluorescent proteins (rsFPs). The available rsFPs for
RESOLFT were generated by few mutations in the sequence of well-known fluorescent proteins such as Dronpa, EGFP, YFP or Cherry.
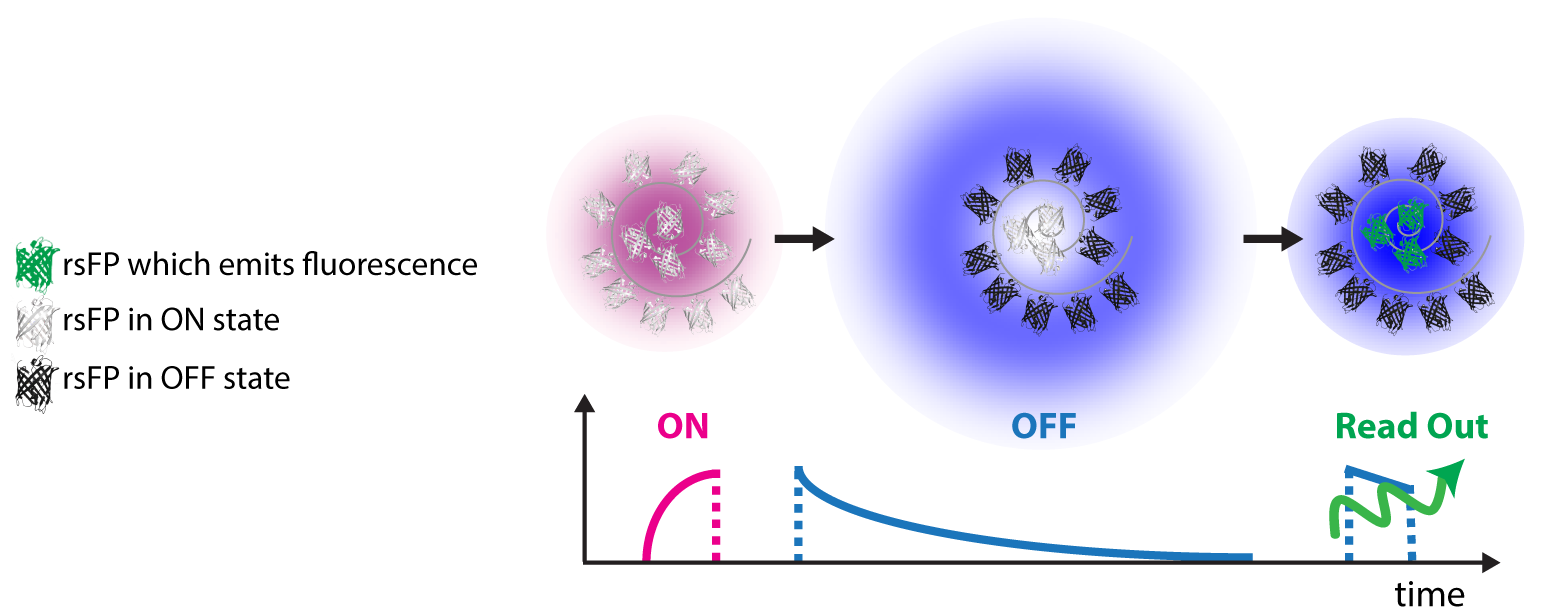
rsFPs can be light driven in different molecular states which inhibit or permit their ability to fluorescence if exposed
to visible light. These states are associated with cis-trans or hydrated-dehydrated molecular configurations. For
RESOLFT to work these states need to be reversibly populated or depopulated multiple times.
The improvements in spatial resolution is in fact achieved by saturating the OFF switching transition using a light
pattern featuring one or more intensity minima or “zeros” (ring or line shaped). The role of this light beam is to
transiently silence the emission of rsFPs by switching them to the long lived off-state. Once switched off, the rsFPs
cannot be excited anymore and remain dark. Only fluorophores residing in the direct vicinity of the zero-intensity
minimum of the RESOLFT light pattern focus can effectively remain into the fluorescent (ON) state and hence contribute
to the fluorescence signal. Importantly, the kinetics of the switching to the OFF state determines the final resolution.
Current point-scanning RESOLFT microscopy equipped with a ring shaped light pattern a lateral resolution down to 30-40nm.
The axial resolution can be improved as well down to 70-90nm by adding an additional light pattern.
This system is comparably cheap and easy to use since it requires only few microwatts of continuous waves
lasers for sample illumination (switching and excitation). Multi-channel variants employing spectral and
lifetime information were also successfully realized enabling colocalization studies at the nanoscale.
This microscope was also upgraded for brain tissue imaging using glycerin objective lens to match the refractive
index of the tissue and additional lenses to correct for spherical aberrations.
A point-scanning implementation of the RESOLFT concept intrinsically compromises the temporal resolution
of the technique, especially in large fields of view where many pixels are recorded. This caveat can be
overcome by implementing parallelized illumination modes to switch and excite the rsFPs. Periodic line patterns
rotated at different angles, as routinely done in structured illumination microscopy, or superimposed at 90
degrees can be successfully implemented to speed up the acquisition of large fields of view in RESOLFT
microscopy. In fact, pioneering studies demonstrated super resolved recordings of a living cell in a field
of view of about 100µm × 100µm happening in few seconds.
STED
Stimulated emission depletion microscopy
In STED microscopy the diffraction barrier is bypassed by controlling the switching of molecules in a deterministic way,
point by point with the superimposition of a light pattern featuring local zero of intensities. STED microscopy uses a
dedicated ‘STED’ beam to modulate the fluorescence capability of fluorophores residing closer than the diffraction
barrier in order to make them distinguishable. In a typical implementation, a focused laser beam is used to raise the
fluorophores to their excited state – as in confocal microscopy. However, the excitation spot is superimposed with the
‘STED beam’, which is typically ring-shaped. The role of this beam is to transiently silence the emission of the fluorophore
by the phenomenon of stimulated emission. Stimulated emission means that the fluorophores are instantly sent to the ground
state before they are capable of emitting a fluorescent photon spontaneously. The silencing takes place because the
wavelength and intensity of the STED beam are chosen so as to take away the majority of the excitation energy in a copy
photon of the STED beam, which is discarded.
Because at least a single de-exciting photon must be available within the lifetime (τ ≈ 1–5 ns) of the fluorescent molecular state, the intensity of the focal STED beam must exceed the threshold Is = Cτ−1 with C accounting for the probability of a STED beam photon to interact with the fluorophore.
Only fluorophores residing in the direct vicinity of the zero-intensity minimum of the STED focus can effectively assume the fluorescent state and hence contribute to the fluorescence signal. If Δ is the conventional resolution (~200-300nm), the diameter of this area is given by
with I
m (typically twice greater than I
s) denoting the intensity at the ring crest.
Hence, features that are (just slightly) more apart than d < λ cannot fluoresce at the same time even
when simultaneously illuminated by excitation light. By scanning the superimposed excitation and ring-shaped
beam focal spots jointly across the specimen, features that are closer than the diffraction barrier assume the
fluorescent state sequentially and are thus readily separated. STED is a direct and immediate imaging method
because it immediately achieves high spatial resolution (down to 15-20nm) without any requirements of image
reconstruction. It is this feature that made STED the method of choice for many applications in cells and even in
living mice.
Smart RESOLFT
Read the paper:
Dreier et al. 2019
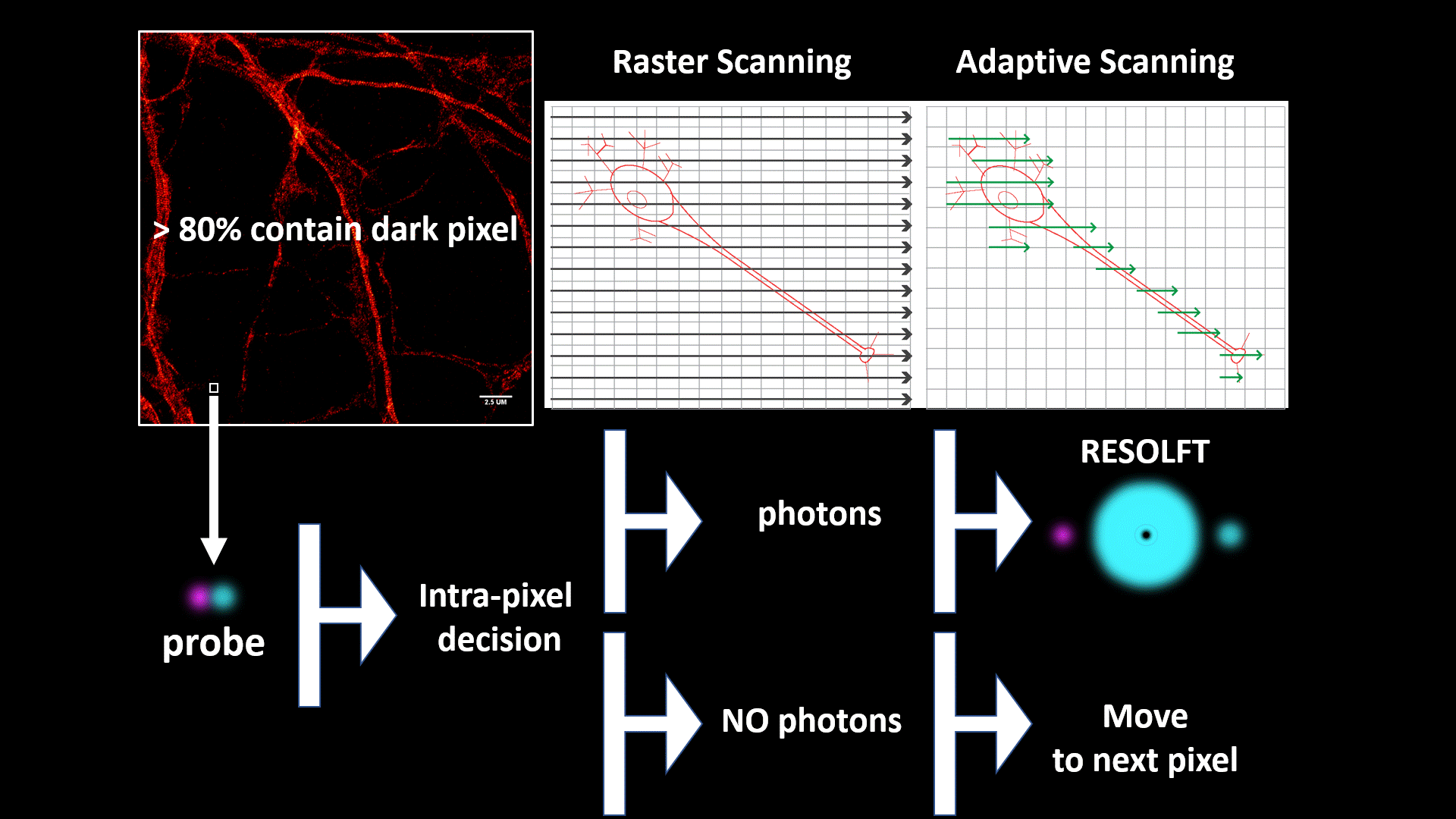
We introduce a novel specimen-adaptive concept of recording super resolution fluorescent images as a step toward a fully automated intelligent microscopy platform.
We applied the concept to RESOLFT optical nanoscopy to perform nanoscale imaging of living cells/tissues/organisms faster and with lower doses of light than previous implementation.
The automated specimen-adaptation is based on the use of a field-programmable-gate-array- (FPGA-)based data-acquisition-card, which enables to apply a rapid feedback loop between illumination, detection and beam scanning. The rapid communications within these modules allows super resolution imaging only in the part of the field of view containing structures without wasting time and energy in dark pixels.
Furthermore, the FPGA allows to classify the signal emitted by the reversible switchable fluorescent proteins during the entire RESOLFT cycle, which provides multiple images of the same structure with increasing spatial resolution – from confocal to RESOLFT - in a single shot.
The information content of these images can be re-assigned and combined in the smart RESOLFT
++ image to increase the image contrast through a custom made multi deconvolution algorithm.
Automated STED
Read the paper:
Alvelid et al. 2020
STED microscopy has been continuously developed in the past decades into a nanoscopy technique that is today used to answer a range of biological questions. Developments of the probes and the optics are both important factors as to why it is one of the most used nanoscopy techniques today, providing a spatial resolution down towards 30 nm in biological samples. What has been lacking is a development in the direction of automation and the possibility to acquire larger FOVs. In our lab, we have developed the technique in this direction, where a combination of optical advancements, additional modules in the microscope, and custom-written hardware control allow our microscope to independently acquire images of theoretically unlimited size.
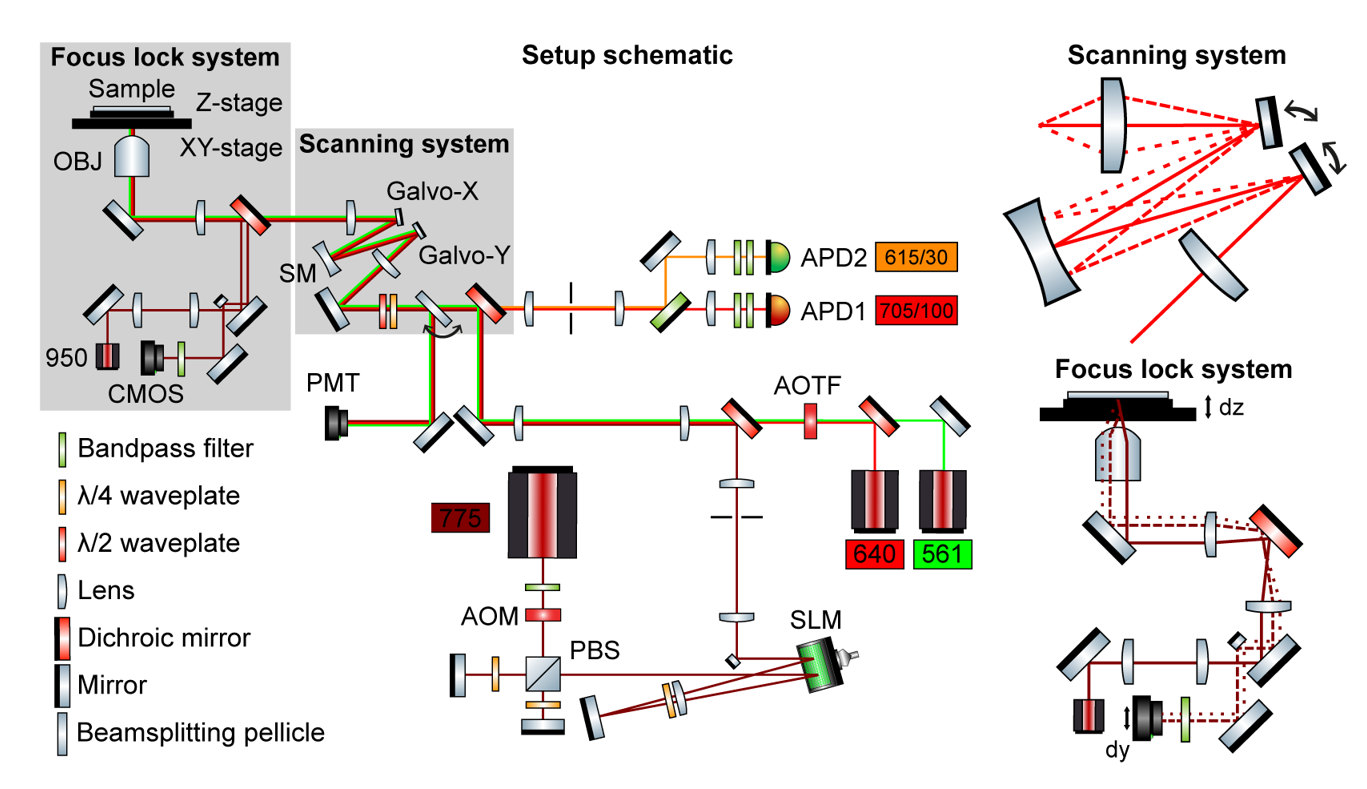
The main optical advancement in the microscope is a scanning system which allows a consistent FOV, both in terms of resolution and intensity, of 80 x 80 µm2. The scanning system consists of two separated galvanometric mirrors, one for each scanning dimension, with a spherical mirror halfway in the optical path between the two. This creates a stationary beam at the back focal plane, no matter the scanning position, and hence both optical aberrations and vignetting are minimized. Furthermore, a focus lock, based on a feedback loop between the axial piezo position and the detected lateral translation of a TIR laser beam as the sample moves along the optical axis, always keeps our sample in focus over prolonged recording times. The custom-written microscope control software that integrates all the parts has successfully allowed the microscope to run without user intervention for overnight recordings. We have shown an imaged area up to 0.53 x 0.53 mm2 in cultured hippocampal neurons.

Custom-written software used to control the microscope is open-source and can be found here.
MoNaLISA
Molecular Nanoscale Live Imaging with Sectioning Ability
Read the paper:
Masullo et al. 2018
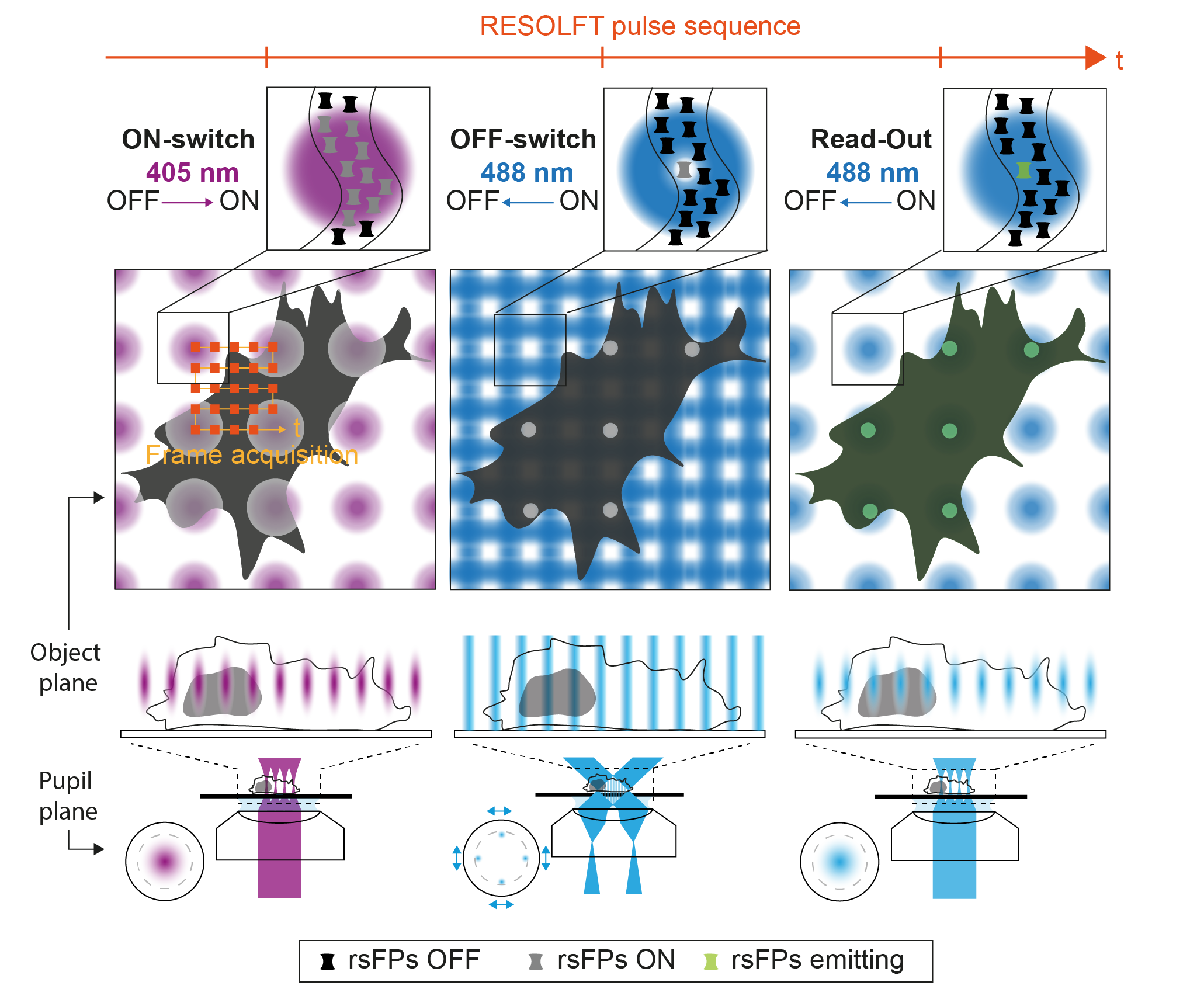
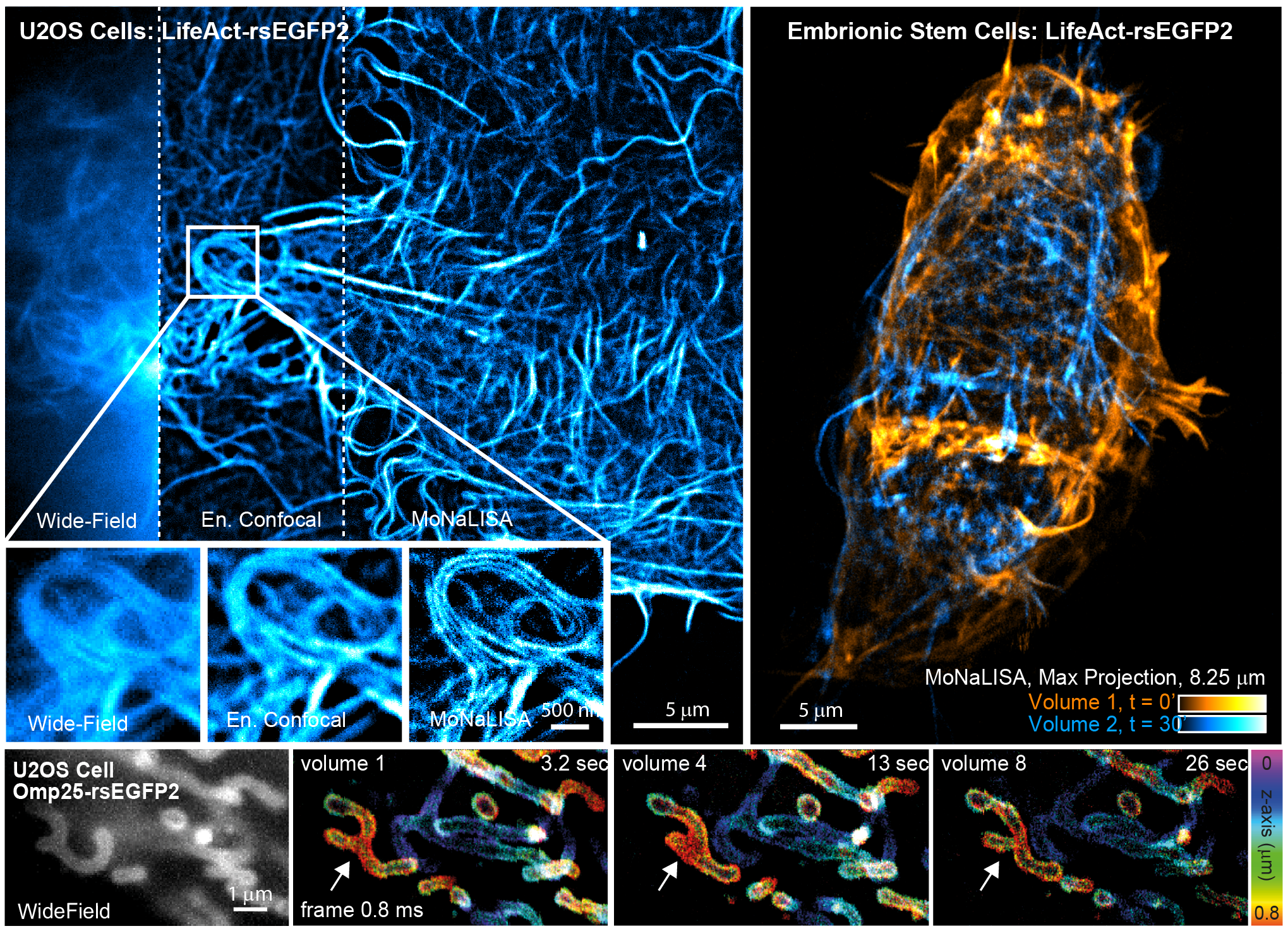
Observing the interplay of organelles and macromolecular complexes inside living cells and tissues calls for the continuous development of minimally invasive optical systems performing at high spatio-temporal resolution. However, the current approaches to fluorescence nanoscopy, even if powerful, are often limited by high doses of light, low contrast, small fields of view or slow recording times. The first limitation can be overcome using Reversible Saturable OpticaL Fluorescent Transition (RESOLFT) approaches, where the coordinate targeted fluorescence ON–OFF switching of reversibly switchable fluorescent protein (rsFPs) requires low light dose (W-kW/cm
2) to obtain sub-100 nm spatial scale. To tackle the other limitations, we developed Molecular Nanoscale Live Imaging with Sectioning Ability (MoNaLISA). The microscope features extended light patterns with optimized shape and periodicity to switch ON, OFF and readout the fluorescence of rsFPs. The OFF-switch illumination is composed of two orthogonal interference patterns which result in periodic vertical and horizontal lines in the image plane. The ON-switch and readout are confined in multispot arrays with larger periodicity in order to maximize the photon collection and minimize switching fatigue and detection cross-talk. This configuration allows to bring optical section in a highly parallelized architecture, giving to the technique the flexibility to visualize structures in the entire cell. The periodic architecture directly translates into speed, releasing the bond between acquisition speed and field of view, and reaching recording of 0.3-1.3Hz with an extended field of view (50x50 μm
2). Furthermore, the fragmentation of the ON-switching in multi-foci leads to a spatially confined protein population that minimize unwanted cycling and photobleaching in areas not directly recorded. As a result, the structure of interest can be visualized for many time points (40-50 frames).
We have demonstrated that MoNaLISA enables 4D imaging at high spatio-temporal resolution and minimal doses of light in several biological systems, from cell to tissues. The examples reported in the figure show actin cytoskeleton structure and dynamics both in adherent cells and in whole embryonic stem cells as they rearrange over time. The speed of the technique can also follow the fine changes in mitochondria morphology during fusion and fission in several sequential volumetric recordings.
3D-pRESOLFT
Parallelized RESOLFT with axial super resolution
Read the paper:
Boden et al. 2021
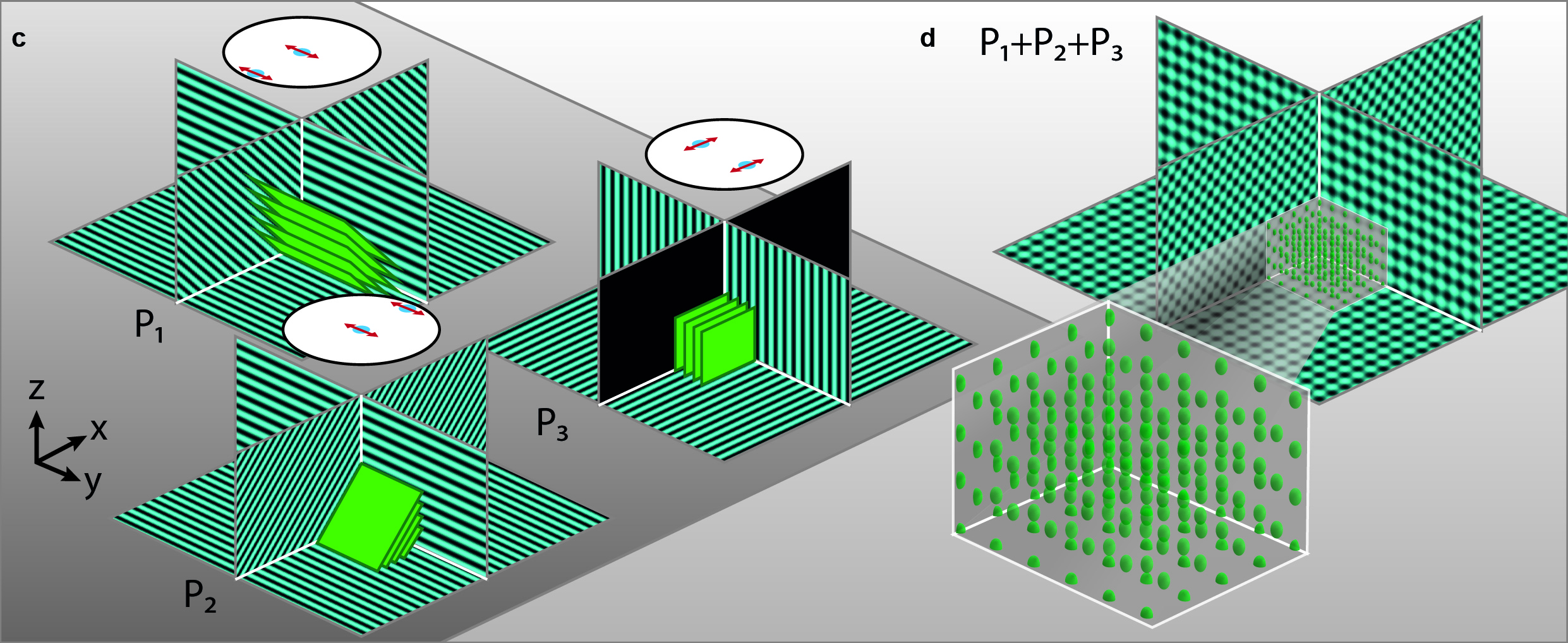
To extend the parallelized RESOLFT concept and achieve super resolution also in the axial direction, we devised a new light pattern for emission confinement. Since axially extended resolution requires axially confined emission, the light pattern used to confine the emission needs to exhibit modulation also in this direction. We achieve this by firstly superimposing two sinusoidal interference patterns that are tilted with respect to the optical axis and finally add a third sinusoidal pattern that is rotated 90 degrees laterally with respect to the first two. Each sinusoidal pattern is created by focusing two coherent beams on the back focal plane of the objective. The tilt is achieved by moving the two focal spots to the side of the back focal plane so that they are not symmetric around the center.
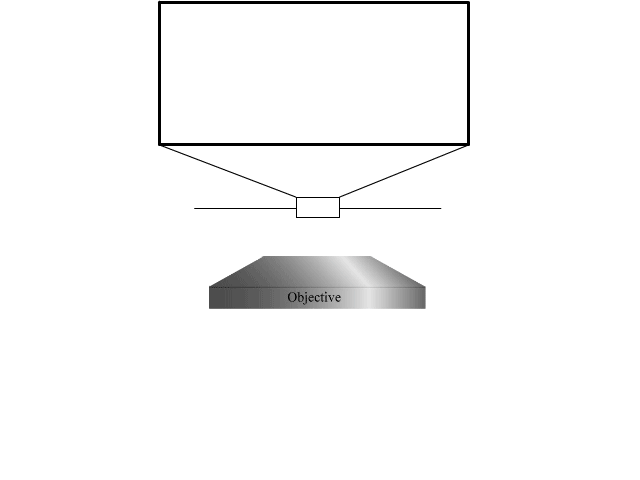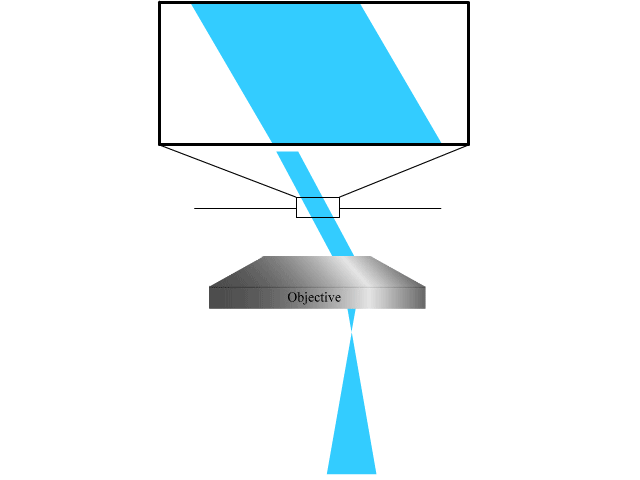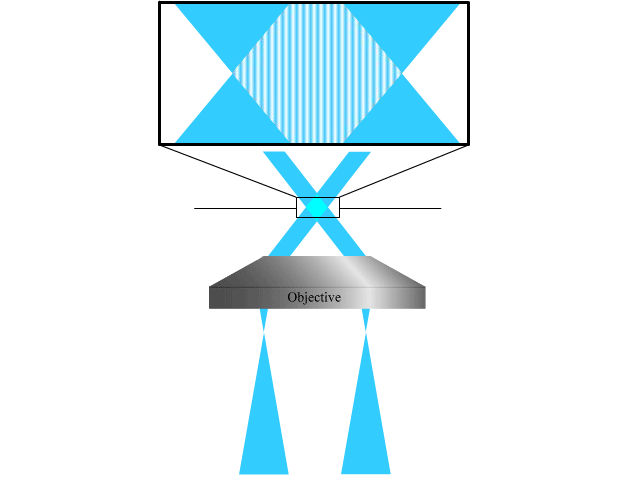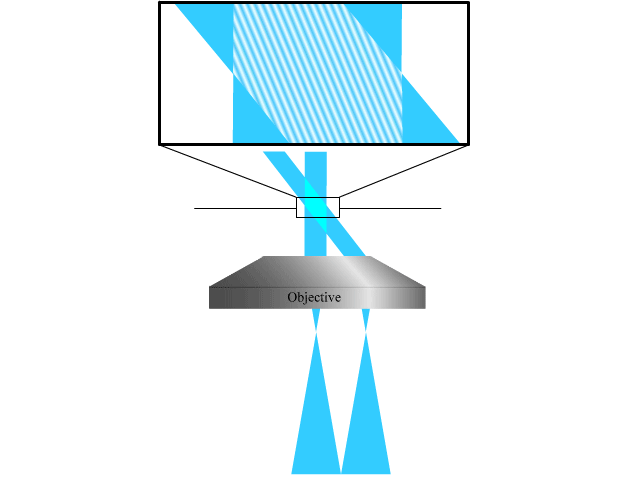
The combination of the three patterns results in a lateral array of three-dimensionally confined zero intensity points. These points are then used with the RESOLFT pulse scheme to achieve three-dimensional parallelized RESOLFT microscopy, or 3D pRESOLFT.